Sunday, October 6, 2024
Applications of Graph Neural Networks (GNNs) in Molecular Structure and Drug Discovery


Introduction to Graph Neural Networks (GNNs)
Graph Neural Networks (GNNs) have emerged as a powerful machine learning framework that extends the capabilities of traditional neural networks to operate on graph-structured data. By leveraging the connectivity and relational information within graphs, GNNs have found applications in a wide range of domains, including social networks, biological data analysis, recommendation systems, and drug discovery.
read more on this here: https://medium.com/@company_56220/introduction-to-graph-theory-and-graph-neural-networks-gnns-dbc4188e42c2
How GNNs Work for Molecular Structures
Graph Neural Networks (GNNs) offer a highly effective way to model molecular structures due to their ability to represent complex relationships between atoms and bonds in chemical compounds. The power of GNNs in this domain lies in how they leverage the inherent connectivity of molecular graphs. A molecule can be viewed as a graph where:
- Nodes represent atoms, each possessing unique properties like atomic number, electronegativity, and charge.
- Edges represent chemical bonds, capturing details like bond order (single, double, triple), bond length, and bond type (covalent, ionic).
Let’s break down how GNNs operate for molecular data:
1. Initialization: Capturing Atomic and Bond Features
The first step in applying GNNs to molecular structures is initializing the graph with the respective features for each atom (node) and bond (edge). These features capture important chemical information, which will be utilized by the GNN to make predictions or generate molecular representations.
Node (Atom) Features
Each atom in a molecule is characterized by several key attributes, which are used as input features for the GNN. These include:
- Atomic number: This identifies the type of atom, distinguishing between elements like hydrogen, oxygen, carbon, etc.
- Electronegativity: A measure of the tendency of an atom to attract electrons in a bond.
- Hybridization state: Describes the orbitals of an atom participating in bonds, such as sp3 for single bonds or sp2 for double bonds.
- Formal charge: The charge assigned to an atom based on its bonding and lone pairs.
Edge (Bond) Features
The bonds between atoms are also initialized with features that describe their chemical properties:
- Bond type: Whether the bond is a single, double, or triple bond.
- Bond length: The distance between two bonded atoms, which can vary depending on the bond type and atomic size.
- Bond polarity: A property that reflects the distribution of electrical charge over the atoms connected by the bond.
These initial node and edge features act as input for the GNN, serving as the foundation for more complex feature extraction and learning during the message passing phase.
2. Message Passing: Learning from Neighboring Atoms
Once the molecular graph is initialized with atom and bond features, GNNs proceed to the message passing phase. This is a core step in which each atom (node) updates its features by aggregating information from its neighboring atoms and the bonds that connect them. Here’s how it works:
Neighborhood Aggregation
In each iteration of message passing, an atom collects information from its directly connected neighbors. This process enables the GNN to gradually capture both local and global structural information. For example, in a molecule like methane (CH₄), the central carbon atom aggregates information from its four connected hydrogen atoms during the first round of message passing. In subsequent rounds, information about the broader molecular structure is also captured.
Each node (atom) updates its feature vector based on the following:
- Features of neighboring nodes: Information about neighboring atoms’ types and properties is aggregated.
- Features of edges: Bond characteristics (like bond type and length) between the node and its neighbors are also considered.
- Custom aggregation function: A function that combines the information from neighboring nodes and edges. Common aggregation functions include summing, averaging, or taking the maximum of the neighbor’s features.
For example, in a molecule with a benzene ring (C₆H₆), an atom in the ring would aggregate information about its adjacent carbon atoms and their respective bonds, capturing the ring’s conjugated system.
Feature Update
After aggregation, the GNN updates the atom’s feature vector using a learned transformation function, often implemented as a neural network layer (e.g., a fully connected layer). This transformation combines the aggregated information with the node’s current features to produce a new, more refined representation.
This process is repeated over multiple iterations (or layers), allowing each atom to incorporate information from farther parts of the molecule. By the end of the message-passing process, each atom’s feature vector encapsulates both its local environment (neighboring atoms and bonds) and more global properties (such as the overall structure of the molecule).
The output of the GNN is a set of updated node and edge representations, which can then be used for a variety of tasks, such as predicting molecular properties or drug-target interactions.
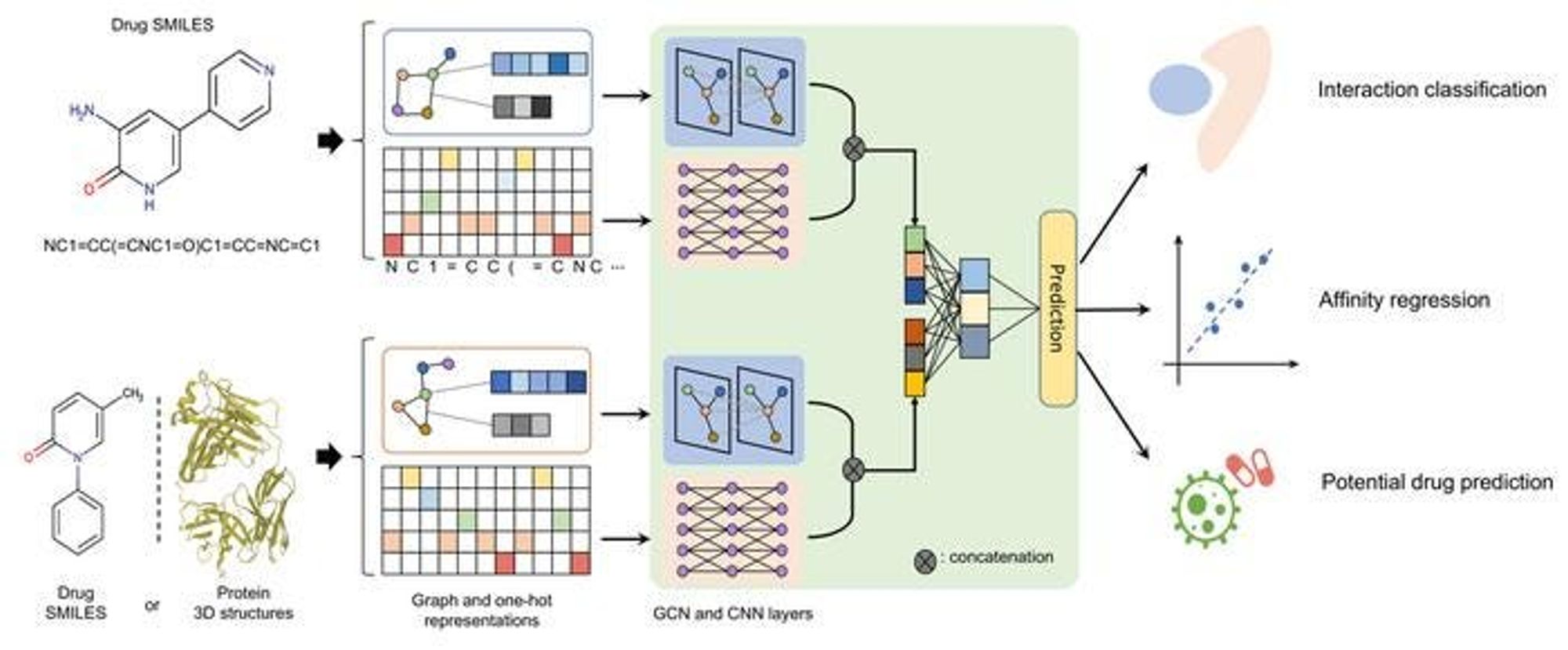
How AI Can Help with Drug Data
The integration of artificial intelligence (AI), particularly through the use of GNNs, has revolutionized the way we handle and interpret drug-related data. Traditional drug discovery processes, which often involve labor-intensive and time-consuming experimental procedures, can now be streamlined and enhanced with AI. Here’s how AI, especially GNNs, is advancing key areas in drug discovery:
1. Predicting Molecular Properties
One of the primary applications of GNNs in drug discovery is the prediction of molecular properties, which can include solubility, toxicity, bioactivity, and stability. By training GNNs on large datasets of molecules with known properties, these models can generalize to predict the properties of new, unseen compounds.
For example, the solubility of a drug molecule, which is crucial for determining its bioavailability, can be predicted by analyzing its molecular structure. GNNs can capture the interaction between atoms in the molecule and predict how these interactions affect the molecule’s overall solubility or toxicity.
By predicting these properties computationally, GNNs reduce the need for exhaustive laboratory experiments, which can be costly and time-consuming. This accelerates the early stages of drug discovery and improves the chances of identifying promising drug candidates.
2. Virtual Screening
Virtual screening is another critical area where AI and GNNs excel. Traditional experimental screening of large chemical libraries to identify compounds with the desired biological effect is laborious. However, AI models can rapidly screen vast libraries to prioritize compounds that are most likely to show the desired activity against a specific biological target.
GNNs model the structural and functional relationships between molecules, making them effective in predicting how a compound will interact with a target protein or biological system. This speeds up the identification of lead compounds, narrowing down the pool of candidates for experimental testing and ultimately saving significant time and resources.
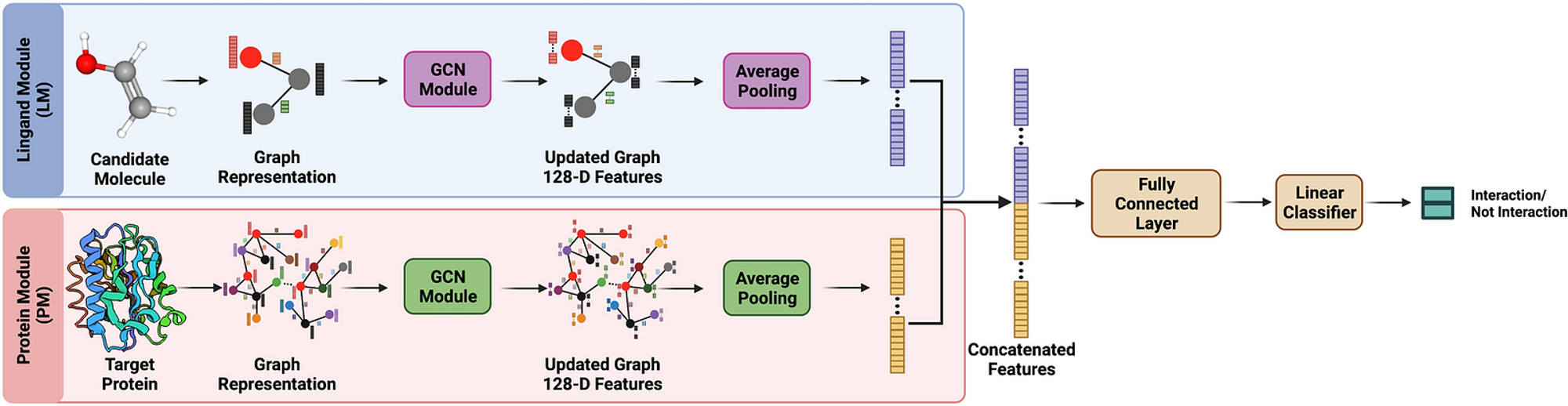
3. Drug-Target Interaction Prediction
Predicting drug-target interactions is one of the most crucial steps in drug development, as it determines whether a drug will bind effectively to its target protein. GNNs can represent both drugs and target proteins as graphs:
- Drug molecules: Nodes are atoms, and edges are bonds.
- Proteins: Nodes are amino acids, and edges represent interactions between these amino acids.
By analyzing the structural similarities and functional relationships within and between these graphs, GNNs can predict binding affinities and interaction sites. This capability helps identify which compounds are likely to bind to a specific target, aiding in the design of more effective drugs with fewer side effects.
4. De Novo Drug Design
One of the most exciting advancements AI brings to drug discovery is de novo drug design, where AI models like Graph Generative Adversarial Networks (GraphGANs) and Variational Autoencoders (VAEs) are used to generate new molecular structures with desired properties.
In this approach, GNN-based models are trained to understand the chemical rules governing molecular formation and then use this knowledge to propose novel compounds. These generative models can ensure that the designed compounds adhere to specific pharmacological and toxicological constraints, such as ensuring that the drug is non-toxic or has high bioavailability.
The AI-generated compounds can then be evaluated and refined, creating an iterative process where new drugs are designed, tested, and improved in a fraction of the time required for traditional methods.
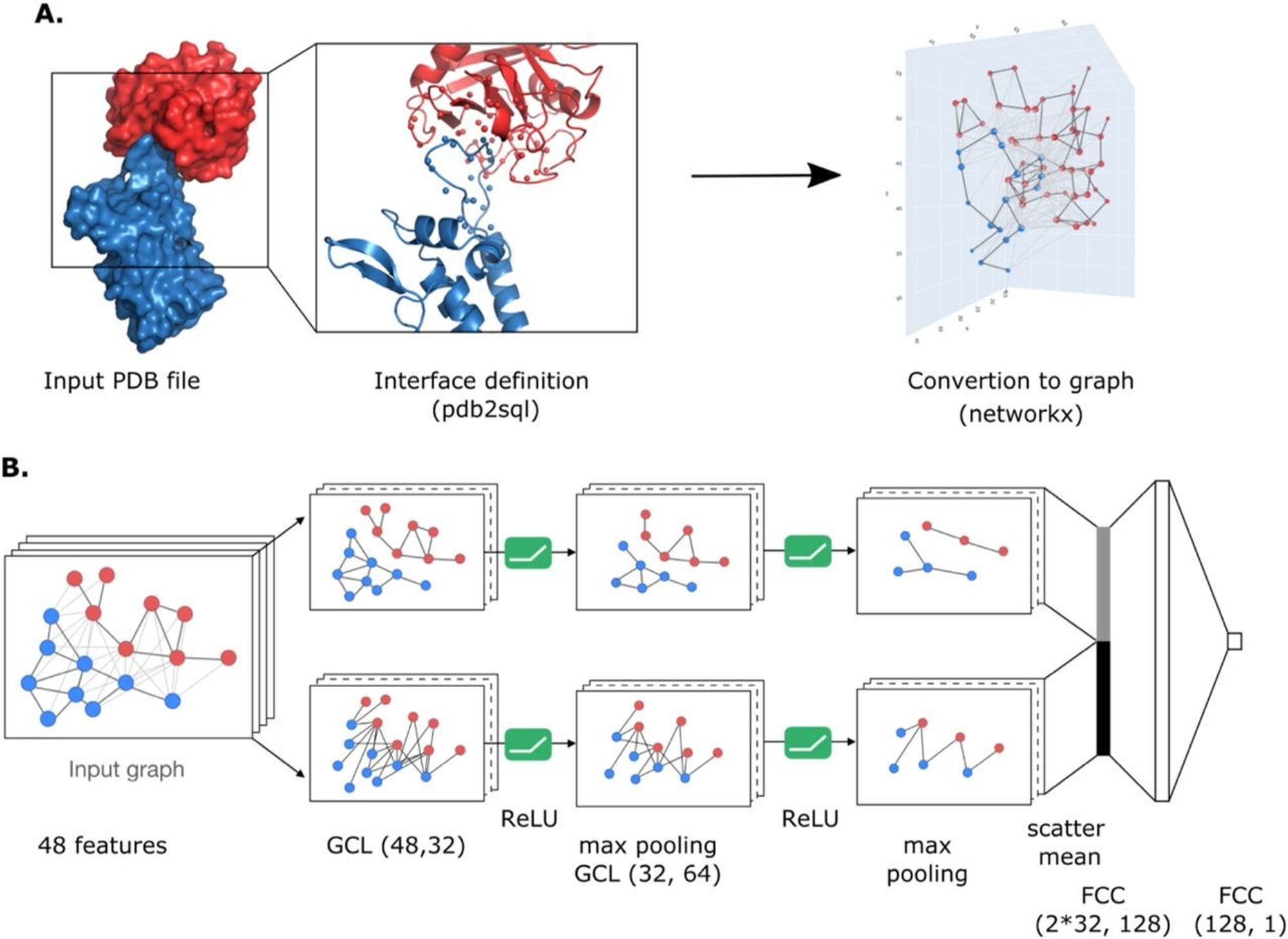
Applications in Drug Working and Discovery
Beyond improving the speed and accuracy of drug discovery, AI and GNNs offer significant advancements in understanding drug mechanisms, tailoring treatments to individuals, and tackling complex diseases.
1. Understanding Drug Mechanisms
AI models can analyze the graphical representation of drugs and provide deep insights into how they interact with biological systems. By modeling the interactions between drugs and their targets at the molecular level, GNNs help elucidate the mechanisms of action, including:
- Binding interactions: Understanding how a drug binds to its target protein.
- Metabolism: Predicting how a drug is metabolized in the body, including identifying potential metabolites and their effects.
- Side effects: By modeling off-target interactions, AI can predict potential side effects and toxicities of drug candidates before they reach clinical trials.
This detailed mechanistic understanding can improve the design of drugs, ensuring that they target the intended biological pathways without unintended consequences.
2. Personalized Medicine
AI’s ability to analyze molecular data extends to personalized medicine, where drug treatments are tailored to individual patients. By combining molecular graph data with patient-specific information, such as genetic profiles and medical history, AI can:
- Predict how an individual might respond to a given drug.
- Identify the most effective drug for that patient.
- Minimize the risk of adverse reactions.
This personalized approach aims to optimize therapeutic efficacy while reducing side effects, moving away from the one-size-fits-all paradigm in medicine.
3. Multi-Target Drug Discovery
Many complex diseases, such as cancer and neurodegenerative disorders, involve multiple biological targets. Designing drugs that interact with multiple targets simultaneously (polypharmacology) can increase their efficacy and reduce the likelihood of drug resistance.
GNNs can model these multi-target interactions, helping researchers design drugs that can tackle multiple pathways at once. This multi-target approach is especially relevant for diseases like cancer, where targeting multiple proteins or pathways can prevent the disease from evolving resistance to treatment.